近日,兰州大学功能有机分子化学国家重点实验室阳铭教授课题组通过从尾至头环化/还原偶联策略实现了trans-clerodanes和sesquiterpene (hydro)quinones的模块化多样性全合成。相关研究成果近期发表在Nat. Commun. 上(DOI: 10.1038/s41467-022-34404-4)。
自然界中存在两种主要的萜类天然产物的生物合成途径,一种是通过聚异戊烯从头至尾的碳正离子环化合成,例如甾体,非甾体类的三萜以及具有十氢萘骨架的萜类分子;另一种是通过聚异戊烯从尾至头的碳正离子环化合成,例如倍半萜天然产物柏木萜烯(funebrene)和cumacrene以及二萜天然产物紫杉烯(taxadiene)。在萜类天然产物仿生合成中,从头至尾环化的合成途径被研究的最广泛,应用也最多。从尾至头的环化仿生合成途径研究的很少,直到2012年才被Scripps研究所的Shenvi课题组首次实现(Nat. Chem. 2012, 4, 915–920)。无论是从头至尾环化的仿生合成还是从尾至头环化的仿生合成,其目的都是为了实现萜类分子的高效合成。而有一类萜类分子,例如trans-clerodanes 和 sesquiterpene (hydro)quinones,虽然他们的生物合成途径都是从头至尾的环化,但是后续又经历了一系列Wagner-Meerwein重排,才得到该类分子的母核骨架(图一)。如果采用从头至尾环化的仿生合成策略来合成该类天然产物,尽管能够迅速实现重排前体十氢萘环骨架的合成,但是其一系列串联的Wagner-Meerwein重排在实验室中很难重现或者需要花费更多的步骤实现。这使得从头至尾环化的仿生合成策略并不能实现该类分子的高效合成。

图一 部分具有相似反式十氢萘环结构的天然产物及其生物合成途径
如果将重排前的中间体12翻转180度之后再和重排后的产物14对比(图二),其反式十氢萘环骨架(蓝色部分)和14的反式十氢萘环骨架(蓝色部分)非常相似。如果能将12中的甲基(红色部分)直接迁移到相应的位置,并且碳正离子消除成双键则可以得到14的反式十氢萘环结构。对比法尼醇衍生物15的Ti(III)促进的开环氧自由基多烯环化的产物16(Tetrahedron 2006, 62, 5215–5222)与trans-clerodanes和sesquiterpene (hydro)quinones的母核骨架,二者也非常相似。不同之处便是16中亚甲基碳的位置和多了一个酯基(红色部分)。而这两部分正是环化前体15中的酯基和甲基(红色部分)转化过来的。如果事先移除15中的酯基和甲基,发生环化之后的产物只需要将羟基转化为相应的甲基则可以从某种意义上实现甲基的“一次迁移”,进而完成trans-clerodanes和sesquiterpene (hydro)quinones的母核骨架的合成。依据这一思路,该课题组设计了环化前体17,如果能顺利环化则会得到产物18,后续将羟基转化为甲基,最后通过大位阻的还原偶联引入不同的侧链就可以实现trans-clerodanes和sesquiterpene (hydro)quinones的模块化多样性合成。与该类天然产物的生物合成途径中从头至尾的环化不一样,该课题组设计的是从尾至头的环化。
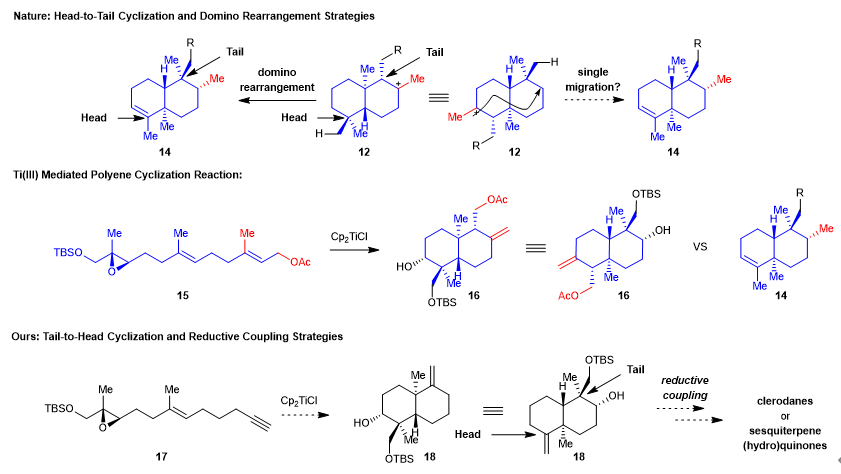
图二 受从尾至头环化/多米诺Wagner-Meerwein重排的生物合成途径启发的从尾至头环化/还原偶联策略合成trans-Clerodanes和Sesquiterpene (Hydro)Quinones
根据这一设计思路,该课题组首先尝试了trans-clerodane天然产物annonene和PL-3的合成(图三)。从醋酸香叶酯的氧化物19出发,Sharpless不对称环氧化引入环氧,并且一锅法保护羟基得到20。铜催化的偶联反应引入炔基侧链,随后一锅法脱除末端炔的三甲基硅基保护基得到环化前体17。尽管当量的Cp2TiCl促进的以炔结尾的开环氧自由基环化已有报导,但是由于当量的Cp2TiCl(2 equiv)开环氧产生的自由基浓度过高,容易与过量的Cp2TiCl反应发生自由基的歧化反应,使得反应体系复杂,因此需要高温来提高环化产物的比例。到目前为止,尚无Cp2TiCl催化的以炔结尾的开环氧自由基环化报导。受Gansäuer课题组关于Cp2TiCl催化的开环氧自由基环化反应的研究(Angew. Chem. Int. Ed. 1998, 37, 101–103)以及Nugent和 RajanBabu关于Cp2TiCl促进的自由基反应的评论性文章(Angew. Chem. Int. Ed. 2021, 60, 2194–2201)的启发,作者在17的环化体系中加入了当量的2,4,6-三甲基吡啶的盐酸盐实现了较低温度下Cp2TiCl催化的环化反应。其中三甲基吡啶的盐酸盐不仅可以作为质子源促进Cp2TiCl2的循环,还可以与体系中的Cp2TiCl络合降低其瞬态浓度,进而抑制自由基的歧化反应的发生。将环化产物18的羟基通过氧化,Wittig反应得到末端烯烃化合物22。TBAF脱除22的硅基保护基,羟基诱导的区域和立体选择性的氢化其中一个末端双键引入所需的甲基,一锅法钴催化内迁另一个末端双键以及羟基溴代得到还原偶联前体23。在兰州大学舒兴中老师开发的还原偶联条件下(Chem. Sci. 2018, 9, 4529–4534)与草酸酯24顺利发生偶联得到天然产物annonene。最后光照氧化呋喃环则得到天然产物PL-3。
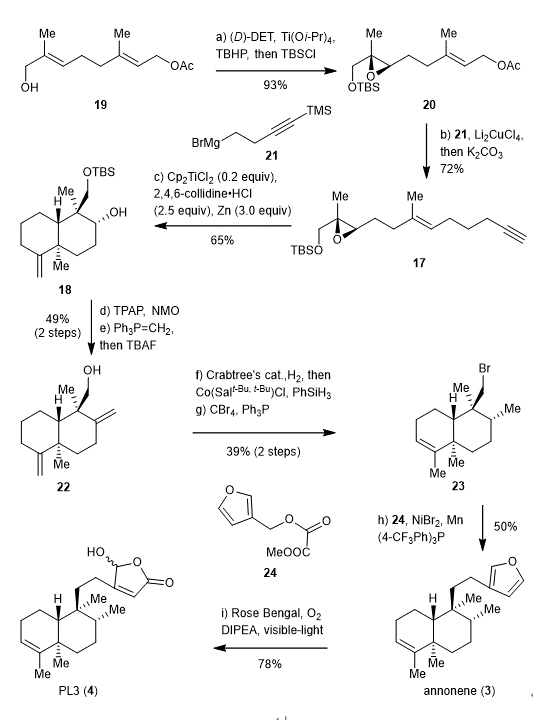
图三 Annonene和PL3的合成
接下来该课题组又将该策略应用到sesquiterpene (hydro)quinones天然产物avarone和avarol的合成中(图四)。由于该类天然产物的母核骨架与trans-clerodane天然产物的母核骨架是对映异构体,因此通过更换Sharpless不对称环氧化所用的手性配体为(R)-酒石酸二乙酯就可以引入相应的原始手性。首先根据前面的路线合成醇25,并将羟基碘代则可以得到还原偶联前体26。随后利用Weix课题组发展的还原偶联条件(J. Am. Chem. Soc. 2012, 134, 6146–6159)顺利的与溴代物27偶联得到共同中间体29。氧化酚到对苯醌就得到天然产物avarone,加还原剂淬灭反应则得到天然产物avarol。
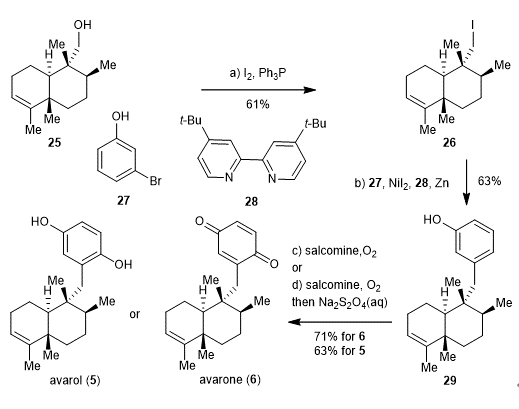
图四 Avarone和Avarol的合成
随后,该课题组又将此策略应用到结构更加复杂的dysidavarone天然产物的合成中(图五)。利用之前同样的路线合成酮32。脱去硅基保护基,将羟基转化为碘代物得到还原偶联前体31。碘代物31与溴代物32发生还原偶联,钯催化的羰基α位芳基化反应得到四环中间体33。通过Wittig反应将羰基转化为末端双键,n-BuSLi脱除酚醚的甲基,把得到的苯酚氧化成对苯醌得到dysidavarones A-C的共同中间体34。氧化的条件下在对苯醌上引入乙氧基得到一对位置异构体—天然产物dysidavarone C和化合物35。Dysidavarone C的末端双键在酸性条件下内迁得到天然产物dysidavarone B,而化合物35在相同的条件下内迁末端双键则得到天然产物dysidavarone A。
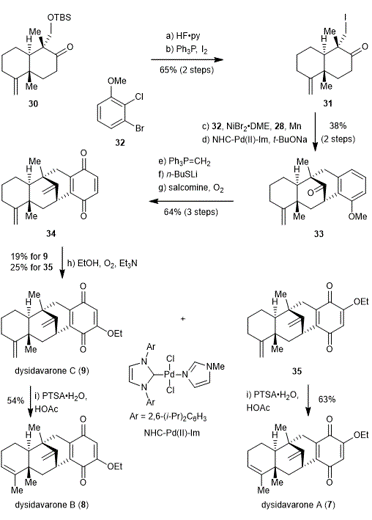
图五 Dysidavarones A-C的合成
Dysidavarone D的结构与A-C不太相同,其C5位的甲基和C9位的甲基处于trans构型。理论上讲,只需要在Sharpless环氧化之前调整相应双键的构型,并且调整环氧化的配体,随后再合成环化前体就能得到dysidavarone D的母核骨架。但是由于开环氧自由基环化反应构象的影响,开环氧后的自由基中间体不直接环化,而是先发生C-C键的旋转以及自由基构型翻转再环化,最终无法得到想要的结构。鉴于此,该课题组决定采用1,3-二羰基化合物的单电子氧化从尾至头的环化策略实现dysidavarone D的母核骨架的合成(图六)。从已知溴代物36出发,通过化合物37的双烯醇负离子烯丙基化反应得到环化前体38。Mn(OAc)3和Cu(OAc)2共同促进的单电子氧化自由基环化反应得到环化产物39。为了选择性还原酯基,作者首先将酮羰基转化为烯醇甲基醚。随后,氢化锂铝还原酯基到醇,用酸淬灭反应的同时将烯醇甲基醚水解得到酮。Appel反应碘代相应的一级醇得到还原偶联前体40。由于碘代物40的C5位的甲基和C9位的碘甲基处于cis构型,其与溴代物32的还原偶联位阻更大,难度也更大。催化量的镍催化剂只能得到极少量偶联产物,而当把镍催化剂和配体28都加大到1.5个当量时,以可以接受的产率得到偶联产物。随后通过钴催化的双键内迁反应,则以两步21%的总产率得到化合物41。通过与之前类似的4步反应得到对苯醌42。虽然直接在醌上引入乙氧基就能得到dysidavarone D,但是引入乙氧基之后的两个位置异构体无法分离,并且引入乙氧基的位置很难确定。因此作者首先将引入乙氧基的醌还原成二酚,然后在位阻小的酚羟基上选择性的引入TBS保护基使两个异构体更容易分离,最后通过NOSEY确定乙氧基的位置之后,脱除TBS保护基并氧化二酚成醌得到合成的dysidavarone D和化合物45。然而合成的dysidavarone D及其异构体45的核磁谱图均与天然样品的核磁谱图不符。这些结果表明天然分离得到的dysidavarone D的结构鉴定有误。
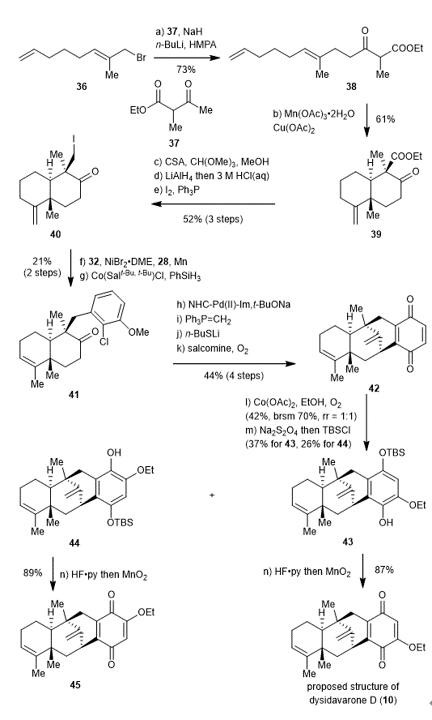
图六 原定结构的dysidavarone D的合成
至此,该课题组采用与从头到尾环化的生物合成途径截然不同的从尾至头的环化策略和还原偶联策略,实现了两个trans-clerodanes天然产物和五个sesquiterpene (hydro)quinones天然产物的模块化多样性全合成。并且通过合成原定结构的dysidavarone D证实了dysidavarone D的结构鉴定有误。
我校博士研究生朱文明为论文的第一作者,博士研究生殷其爽为共同第一作者,萃英学院本科生娄之正参与了该研究工作。
该工作受到国家自然科学基金青年基金和面上项目,兰州大学,中央高校基本科研业务费和功能有机分子化学国家重点实验室的大力资助。