核苷类抗生素A201A、A201E、A201D由Eli Lilly公司在1976年从链霉菌Streptomyces capreolus NRRL 3817 中分离得到的,对革兰氏阳性菌和大部分厌氧性革兰氏阴性菌表现出极佳的抗菌活性。作用机理是与核糖体结合,抑制mRNA的翻译从而抑制蛋白质的合成。2012年,鞠建华课题组从南中国海海底获取的放线菌株中又意外地分离得到了A201A、A201E 及类似物。
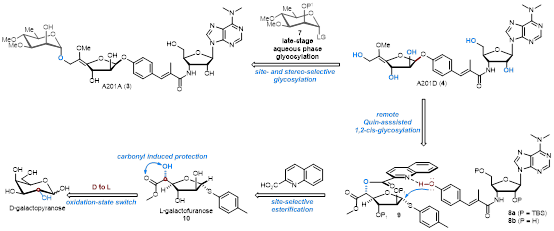
近日兰州大学王晓磊课题报道了A201A、A201E、A201D的全合成研究。其关键骨架由C1和C6头尾氧化态转换的策略实现:即从便宜易得的D-吡喃型半乳糖实现了L-呋喃型半乳糖供体的合成;随后,又通过2-喹啉甲酰远程导向的糖苷化,构筑了A201家族中具有挑战性的1,2-顺式呋喃糖苷核心结构;最后,又在水相中立体选择性的引入了D-鼠李糖或俞氏糖苷化,实现了A201A和A201E的全合成。该合成路线简洁高效,为后期A201家族的构效关系研究奠定了基础。